醫療器械作為直接或間接用于人體的儀器、設備、器具,其安全性、有效性至關重要。產品技術要求是醫療器械研發、生產、檢驗和監管的核心文件,它詳細規定了產品的性能指標、檢驗方法及質量保證要求。為了讓相關從業者及公眾更直觀地理解其核心內容,以下將結合圖表進行解析。
一、 技術要求的主要內容結構
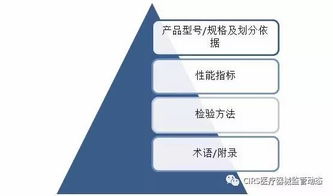
一個完整的技術要求文件通常包含以下幾個核心部分,其結構關系如下圖所示:
graph TD
A[醫療器械產品技術要求] --> B1[產品型號/規格及其劃分說明]
A --> B2[性能指標]
A --> B3[檢驗方法]
A --> B4[附錄(如有)]
B2 --> C1[物理性能]
B2 --> C2[化學性能]
B2 --> C3[電氣安全性能]
B2 --> C4[生物性能]
B2 --> C5[軟件功能]
B3 --> D1[方法原理]
B3 --> D2[儀器與試劑]
B3 --> D3[試驗步驟]
B3 --> D4[結果判定]二、 核心要求詳解
- 性能指標:這是技術要求的核心,必須具體、可測量。
- 物理性能:如尺寸、重量、強度、硬度、疲勞度等。例如,骨科植入物的尺寸公差、注射器的活塞滑動性能。
- 化學性能:如材料成分、重金屬含量、酸堿度、殘留溶劑、可瀝濾物等。這對接觸人體體液或組織的器械尤為重要。
- 電氣安全與電磁兼容:對于有源器械,必須符合GB 9706.1(醫用電氣設備安全通用要求)等標準,確保防電擊、防機械危險、輻射安全等,且工作時不對其他設備產生干擾,也能抵抗外界干擾。
- 生物性能:依據器械與人體接觸的性質和時間,需進行細胞毒性、致敏、刺激、全身毒性等生物學評價,確保生物相容性。
- 軟件功能:對于含軟件的器械(如診斷設備、治療計劃系統),需明確軟件的功能、運行環境、用戶界面、數據接口及網絡安全要求。
- 檢驗方法:每一項性能指標都必須有對應的、科學公認的檢驗方法。方法需明確到足以讓不同實驗室復現相同結果,通常引用國家標準(GB)、行業標準(YY)或國際標準(如ISO、IEC)。
- 型號規格劃分:清晰界定同一注冊單元下不同產品的區別(如不同尺寸、配置),并說明其性能指標的共性與差異。
三、 技術要求制定與監管流程
技術要求并非一成不變,其生命周期與產品研發和監管緊密相連。
graph LR
E[產品設計與研發] -- 依據預期用途與法規標準 --> F[起草技術要求草案]
F -- 內部驗證與檢測 --> G{指標與方法是否<br>科學、可測、可控?}
G -- 否 --> F
G -- 是 --> H[注冊檢驗與產品定型]
H --> I[提交注冊審批]
I --> J[國家藥監局技術審評]
J --> K{是否批準?}
K -- 否,需補正 --> F
K -- 是 --> L[獲準注冊,技術要求成為法定文件]
L --> M[生產過程嚴格依標控制]
M --> N[上市后監管與變更管理]
N -- 產品升級/工藝變更 --> F四、 關鍵要點與意義
- 法規符合性:技術要求是《醫療器械監督管理條例》的法定要求,是產品注冊的必備文件。
- 質量基石:它是企業組織生產、進行質量控制的根本依據,確保產品批次間的一致性。
- 監管標尺:是藥品監督管理部門開展上市后監督抽檢、不良事件調查的判斷基準。
- 動態發展:隨著科技進步、標準更新或臨床反饋,技術要求可通過變更注冊進行修訂和完善。
醫療器械產品技術要求是將法規、標準、科學與具體產品相結合的結晶。通過清晰的圖表化解讀,可以幫助研發人員精準設計,生產者規范制造,監管者有效監督,最終共同守護公眾用械安全。